What Depression Actually Does to the Brain, and Why Most Explanations Stop Too Early
Depression is one of the most misunderstood neurological states in modern practice. Not because the research is lacking, it isn’t, but because the popular narrative around this condition stopped evolving roughly twenty years ago. Serotonin deficiency became the default explanation. SSRIs became the default response. And for the individuals who came to me having already tried every standard protocol without lasting change, that explanation was worse than incomplete. It was actively misleading.
The serotonin hypothesis has been quietly losing ground in the research literature for years. A landmark 2022 umbrella review published in Molecular Psychiatry examined the evidence across six domains and found no consistent support for the idea that low serotonin causes this condition. What the research does support, with increasing specificity, is that depression involves significant disruption to the dopaminergic system: the brain’s motivation, anticipation, and reward architecture. Understanding that distinction changes everything about how it can be addressed.
This content explores the neuroscience of depression. For personalized assessment, contact MindLAB Neuroscience directly.
The Dopaminergic Underactivation Signature of Depression
When I work with individuals carrying this presentation, the neurological signature I observe most consistently is not emotional numbness in the traditional sense. It is dopaminergic underactivation: a measurable reduction in the brain’s capacity to generate anticipatory reward signals. The ventral tegmental area, nucleus accumbens, and prefrontal cortex, the core nodes of the mesocorticolimbic dopamine circuit, show suppressed activity. The result is not simply sadness. It is the architectural collapse of wanting.
This is why antidepressants targeting serotonin reuptake frequently produce only partial remission. They address mood regulation circuits without touching the motivational deficit at the center of the experience. In research terms, nucleus accumbens hypoactivity observed in major depression predicts anhedonia severity far more reliably than serotonergic measures do: a finding consistent with the broader science of dopamine and motivational drive architecture. That is a critical clinical finding, and one that rarely reaches the individuals most affected by it.
In over two decades of practice, I have seen this pattern consistently: individuals who describe their state not as profound sadness but as a complete absence of forward pull. Nothing draws them toward tomorrow. Goals that once felt meaningful feel hollow. This is the dopaminergic signature of depression, and it requires a fundamentally different intervention than mood stabilization alone.
Anhedonia and Sadness: Two Separate Neural Circuits Inside the Same Presentation
One of the most important distinctions in this field, one that standard explanations routinely collapse, is the difference between anhedonia and sadness. These are not different intensities of the same experience. They involve different neural circuits, different neurochemical profiles, and different intervention targets.
Sadness is processed primarily through the limbic system, involving the amygdala and its connections to the anterior cingulate cortex. It is an emotional response with neural correlates that overlap substantially with grief, loss, and social pain. Uncomfortable, yes, but also, neurologically speaking, a signal. The brain’s way of registering meaningful loss and prompting reassessment.
Anhedonia is something categorically different. Anhedonia, the inability to experience pleasure or anticipate reward, operates through dopaminergic circuits, not primarily limbic ones. The ventral striatum, which normally activates in anticipation of pleasurable experiences, shows markedly reduced responsivity in individuals with depression who present with anhedonia. The brain is not generating the forward-pull signal that normally motivates behavior. This is why anhedonia, far more than sadness, predicts functional impairment, and why individuals experiencing it often report feeling confused by their own state. They are not in emotional pain in the way the word “depressed” implies. They are in a motivational void.
When depression is treated as exclusively a sadness problem, anhedonia goes unaddressed. The mood may stabilize. The motivational flatness remains. And individuals (particularly those who are otherwise functional, high-achieving, and analytically capable) conclude that something is fundamentally wrong with them rather than recognizing that the intervention was aimed at the wrong circuit.
The High-Performer Depression Paradox: Functional Outside, Depleted Inside
The most frequently misidentified form of depression I encounter does not look like what most people imagine. The individual experiencing it is still showing up. Still producing. Still maintaining the relational and professional structure of a life that, from the outside, appears intact. What the outside cannot see is the neural exhaustion required to sustain that output when the dopaminergic reward system has gone quiet.
I have worked with executives, founders, and high-functioning professionals who came to me after years of carrying this exact paradox. They were not lying in bed unable to function. They were running companies, managing complex relationships, and by every external metric doing well. Internally, they described a state consistent with the neurological profile of this condition: the absence of genuine anticipation, the mechanical execution of previously meaningful work, the inability to convert achievement into any lasting sense of satisfaction.
This is what researchers increasingly describe as high-functioning depression: a presentation where compensatory mechanisms and learned high-performance habits mask the underlying neurological deficit. The behavioral scaffolding holds up. The dopaminergic architecture does not. And the longer the gap between external performance and internal depletion, the more brittle the scaffolding becomes.
The danger of misidentifying this pattern is significant. This condition in high performers often goes unaddressed for years precisely because the external functioning appears intact. The individual does not fit the culturally familiar image of what this condition looks like. The gap between presentation and internal experience is itself a hallmark of how it manifests in neurologically capable, cognitively driven individuals, and it demands a different analytical lens entirely.
Neuroinflammation and the Hypothesis That Changes the Intervention Calculus
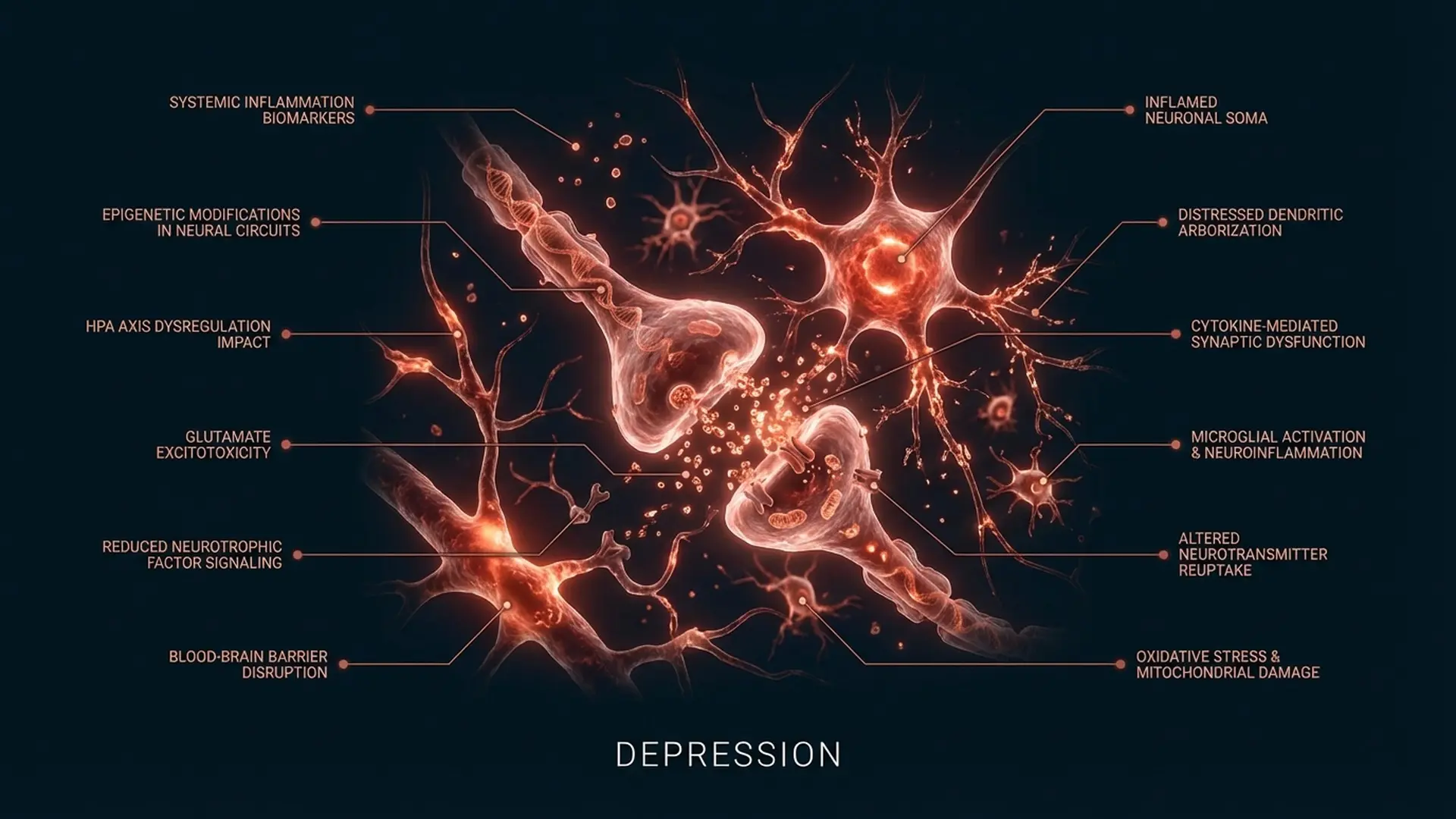
The neuroscience of depression has expanded substantially in the last decade, and the evidence for a neuroinflammatory component has become impossible to dismiss. Elevated levels of pro-inflammatory cytokines (particularly IL-6, TNF-alpha, and CRP) are consistently observed in individuals with this condition, and the correlation is not peripheral. Neuroinflammation disrupts the very circuitry that regulates motivation, reward, and threat calibration.
Specifically, inflammatory signaling interferes with tryptophan metabolism, reduces synaptic serotonin availability, and, critically, disrupts the mesolimbic dopamine pathway. Cytokines have been shown to reduce tyrosine hydroxylase expression, the enzyme required for dopamine synthesis. They also affect the striatum’s sensitivity to dopamine signals. The neuroinflammatory hypothesis, in other words, does not sit separately from the dopaminergic underactivation signature of depression: it helps explain the mechanism.
For intervention purposes, this matters enormously. A neurologically depleted state driven by chronic inflammatory load is not primarily a thought pattern problem. It is not a cognitive distortion problem. It is a physiological disruption with downstream effects on motivation, reward, and threat sensitivity. Approaches that exclusively target thought patterns (reframing, cognitive restructuring, behavioral activation in isolation) are aiming at consequences rather than the underlying mechanism. In my practice, I routinely see this: individuals who have done extraordinary amounts of cognitive work on their depression without the motivational depletion resolving, because the intervention was never aimed at the dopaminergic architecture or the inflammatory variables affecting it.
Why Standard Approaches Miss the Motivational Core
Standard approaches are designed around the assumption that depression is primarily a mood state problem. That assumption shapes everything: the clinical classification criteria, the pharmacological targets, the therapeutic modalities, and the outcome measures. When this condition presents primarily as motivational collapse rather than dysphoric mood, the standard framework consistently underserves the individual.
Consider how the motivationally dominant presentation is typically experienced: the individual wakes not feeling sad but feeling nothing. The things that previously created forward momentum (work, connection, creative pursuit, physical activity) have lost their pull. Not their value, intellectually. Their pull, neurologically. The mesolimbic reward signal that would normally generate anticipatory arousal around these activities has gone quiet. What remains is a state of effortful execution without genuine engagement.
Standard cognitive approaches attempt to address this by restructuring the thoughts associated with motivational deficits. Behavioral activation protocols attempt to initiate behavior in the hope that the dopaminergic response will follow. These approaches are not without value. But they are working upstream of the actual neurological problem. The dopaminergic circuit that generates wanting must itself be the intervention target, not just the behaviors that depend on it.
I have seen individuals spend years in otherwise competent therapeutic work, accumulating insight into the patterns associated with their depression, without the fundamental motivational deficit shifting. The insight is real. The behavioral awareness is real. The neural architecture generating the anhedonia and motivational flatness has not been touched. This gap, between psychological insight and neurological change, is precisely where the standard approach runs out of leverage.
Dr. Ceruto’s Dopaminergic Recalibration Methodology
My approach is built on a foundational premise that distinguishes it from standard frameworks: depression is not a mood to be managed but a neural architecture to be recalibrated. Specifically, the dopaminergic circuitry governing anticipation, reward, and motivational drive must be the direct target of intervention, not a secondary beneficiary of mood stabilization.
The first phase of this work involves precise identification of the neurological signature the individual is actually experiencing. Is the dominant presentation anhedonic: the motivational void, or dysphoric: the active suffering? Is there evidence of neuroinflammatory load: chronic sleep disruption, elevated inflammatory markers, patterns of high allostatic burden? Is this a high-performer presentation where compensatory functioning is masking the depth of the depletion? These distinctions determine the intervention architecture.
The second phase targets the dopaminergic reward circuit directly. Real-Time Neuroplasticity™ works within the live moment: identifying the precise neural signals the brain is using to suppress or bypass anticipatory reward generation, and intervening in real time to begin restructuring those patterns. This is not retrospective analysis. It is engagement with the actual neural event as it occurs, in the contexts where the grip of depression is most active. The plasticity window that opens during genuine high-stakes engagement is narrower and more powerful than anything accessible in a reflective, retrospective frame.
What I consistently observe when this methodology is applied correctly is not a gradual mood improvement. It is a restoration of the forward-pull signal: the return of genuine anticipation, the reactivation of the mesolimbic reward response around the activities and relationships that matter to the individual. This is neurologically distinguishable from mood management. It is the circuit itself beginning to function differently.
The Next Step: Identifying the Neurological Profile Driving Your Experience
Depression is not one experience. It is not one mechanism. And it is not one intervention. The research is clear that the dopaminergic and neuroinflammatory components require targeted engagement: engagement that goes beyond the serotonergic and cognitive frameworks that still dominate standard practice. The individuals who come to MindLAB Neuroscience typically arrive after years of working within those frameworks, often with significant insight and real progress, but without the fundamental neurological shift that resolves the motivational depletion at the center of their experience.
The distinction between anhedonia and sadness matters clinically. The high-performer presentation of depression, where external functioning masks internal depletion, matters clinically. The role of dopamine system dysregulation in motivational collapse matters clinically. The neuroinflammatory dimension matters clinically. And the intervention target (the dopaminergic architecture, not just the associated mood states or thought patterns) matters clinically. Understanding which mechanisms are driving this condition is the necessary precondition for identifying what can actually change it.
This content explores the neuroscience of depression. For personalized assessment, contact MindLAB Neuroscience directly. To explore whether the Real-Time Neuroplasticity™ methodology is the appropriate intervention for your specific neurological profile, schedule a strategy call with Dr. Ceruto.